Sie sind hier: Startseite > Hoden > Intersexualität > Adrenogenitales Syndrom
Ursachen und Therapie des adrenogenitalen Syndroms (AGS)
Definitionen des adrenogenitalen Syndroms
Das adrenogenitale Syndrom ist ein autosomal rezessiv vererbter Enzymdefekt der Steroidbiosynthese, welche zu einer exzessiven ACTH-Erhöhung aufgrund des Kortisolmangels führt (Merke und Bornstein, 2005). Die Steroidzwischenprodukte vor dem Enzymdefekt sind exzessiv erhöht und führen zu einer Nebennierenhyperplasie (engl. Krankheitsbezeichnung: congenital adrenal hyperplasia, CAH). Siehe auch Abb. Steroidbiosynthese. Der zugrundeliegende Enzymdefekt und das Geschlecht des Patienten bestimmen die Symptome.
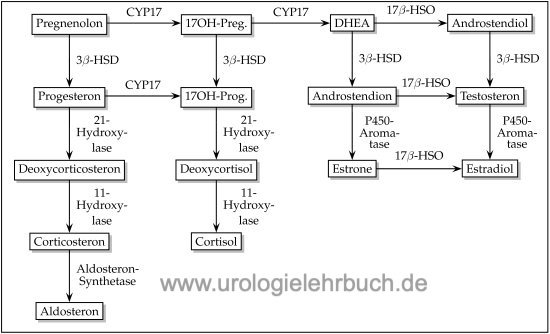 |
Ursache und Klinik des adrenogenitalen Syndroms
21-Hydroxylase-Defekt:
Der häufigste Enzymdefekt (95%) für ein adrenogenitales Syndrom mit einer Inzidenz von 1:14 000 Geburten.
Es gibt zwei klinische Verlaufsformen je nach Schwere des Enzymdefektes: Virilisierung oder Virilisierung mit Salzverlust aufgrund einer verminderten Aldosteronsynthese.
21-Hydroxylase-Defekt ohne Salzverlust:
bei Mädchen entsteht je nach Schwere eine milde Virilisierung (normale weibliche Genitale bei Geburt mit späterer Klitorisvergrößerung und Hirsutismus) oder schwere Virilisierung (Maskulinisierung der Genitalien bei Geburt).
Jungen fallen klinisch zunächst nicht auf, später schnelles Wachstum, frühe Pubertät, kleine Hoden, früher Wachstumsstop.
21-Hydroxylase-Defekt mit Salzverlust:
durch die verhinderte Aldosteron- und Kortisonsynthese entsteht ein Flüssigkeits- und Salzverlust ab dem 5. Lebenstag. Hyponatriämie und Hyperkaliämie sind typisch, weiterhin Azidose, Dehydration, Maskulinisierung bei weiblichen Neugeborenen, große Genitalien bei männlichen Neugeborenen.
Diagnose des 21-Hydroxylase-Defekts:
Erhöhtes 17-Hydroxyprogesteron, Androgene (Androstendion, Dehydroepiandrosteron oder Testosteron) und ACTH. Erniedrigtes Kortison, bei Salzverlust ist auch Aldosteron vermindert trotz erhöhter Reninaktivität.
11beta-Hydroxylase-Defekt:
für 5 % der adrenogenitalen Syndrome verantwortlich. Durch den Enzymdefekt wird die Kortisolsynthese verhindert, dies verursacht stark erhöhtes 11-Desoxycorticosteron und 11-Desoxycortisol. Alle Patienten haben einen Hypertonus, ausgeprägter Enzymdefekte führen auch zu erhöhten adrenalen Androgenen und zu Virilisierung.
Seltene Enzymdefekte des adrenogenitalen Syndroms:
Defekte der 3beta-Hydroxysteroid Dehydrogenase, 17alpha-Hydroxylase, und StAR (steroidogenic acute regulatory protein) führen auch zu einer gestörten Testosteronbiosynthese:
StAR-Mangel
Mutationen im steroidogenic acute regulatory protein (StAR) führt zum fehlenden Transport von Cholesterol durch das Mitochondrium und damit zum Mangel an allen Steroiden. Bei männlichen Betroffenen entstehen weibliche äußere Genitale mit blind endender Vagina ohne Uterus. Große fettgeladene Nebennieren verdrängen die Nieren nach kaudal. Bei weiblichen Betroffenen sind die Genitalorgane normal. Eine ausgeprägte Nebennierenrindeninsuffizienz kann bei beiden Geschlechtern postnatal zum Tode führen. Für die Therapie ist die Hormonsubstitution essentiell.
3β-Hydroxysteroid Dehydrogenase-Mangel
Seltene Form des adrenogenitalen Syndroms. Der Enzymdefekt führt zur fehlenden Umwandlung von Pregnenolon in Progesteron, DHEA in Androstendion, Androstendiol in Testosteron. Es gibt verschiedene Ausprägungen des Enzymdefekts. Bei der klassischen (ausgeprägten) Form führt der Mangel an Aldosteron, Kortisol, Testosteron und Estradiol zu Salzverlust, Virilisierung der weiblichen Patienten und fehlender Virilisierung der männlichen Patienten. Mildere Formen verursachen keinen Salzverlust, das klinische Bild ist unspezifisch (u.a. Hypospadie und Mikropenis bei Jungen, bei Mädchen Hirsutismus und Infertilität).
17α-Hydroxylase-Mangel
Seltene Form des adrenogenitalen Syndroms. Der Enzymdefekt führt zur fehlenden Bildung von 17–Hydroxyprogesteron und 17–Hydroxypregnenolon, in der Konsequenz eines Mangels an Kortison und Sexualsteroiden. Es entsteht exzessiv Desoxycorticosteron und Folgeprodukte mit Hypertonie, Hypokaliämie und Alkalose.
Das männliche Geschlecht hat weibliche oder uneindeutige äußere Geschlechtsmerkmale, das weibliche Geschlecht normale Genitalien. Bei weiblichen Patienten bleibt die Pubertät aus. Beide Geschlechter entwickeln einen Hypertonus.
Testikuläre adrenale Resttumoren (TART):
Versprengte Nebennierenzellen im Hoden proliferieren unter Einfluss von ACTH, insbesondere bei schlechter medikamentöser Einstellung. Es enstehen Infertilität (bis zur Azoospermie) und tastbare Hodentumoren, welche jedoch benigne sind. Eine supranormale Subtitution mit Kortison führt zur einer deutlichen Rückbildung der testikulären adrenalen Resttumoren. Der Tastbefund und die Sonographie kann TART nicht von Keimzelltumoren unterscheiden, wegweisend ist der bilaterale Befund und das Ansprechen auf Kortison. Eine operative Freilegung sollte vermieden werden.
Therapie des adrenogenitalen Syndroms
Die Akuttherapie der Addison-Krise beinhaltet Kortisongaben sowie Flüssigkeits- und Elektrolytsubstitution. Im weiteren Verlauf ist die lebenslange Substitution von Kortison und Fludrokortison notwendig. Die Dosierung hängt von den Hormonkonzentrationen, den Elektrolyten, dem Blutdruck, dem Wohlbefinden und Wachstum des Patienten und Zeichen der Virilisierung ab. Bei ausgeprägter Maskulinisierung können frühzeitige Korrekturoperationen wie z.B. eine Reduktion der Klitoris durchgeführt werden, dies ist jedoch bei fehlender Einwilligungsfähigkeit des Kindes umstritten. Für die Fertilität von betroffenen Männern ist eine korrekte Hormoneinstellung essentiell.
| Weibl. Pseudohermaphroditismus | suchen | Männl. Pseudohermaphroditismus |
Sachregistersuche: A B C D E F G H I J K L M N O P Q R S T U V W X Y Z
Literatur
C. Radmayr, G. Bogaert, H. S. Dogan, and Tekgü, “EAU Guidelines: Paediatric Urology,” 2022. [Online]. Available: https://uroweb.org/guidelines/paediatric-urology/.
 English Version: Enzyme defects of congenital adrenal hyperplasia and treatment
English Version: Enzyme defects of congenital adrenal hyperplasia and treatment
Urologielehrbuch.de ohne Werbung
Diese Internetseite ermöglicht mit Hilfe von Werbung den Volltext-Zugriff auf das aktuelle Urologielehrbuch.de. Viele Bilder sind zum Schutz von Laien verpixelt oder ausgeblendet. Regelmäßig wiederkehrende (fachkundige) Leser können die Werbebanner abschalten und Zugriff auf alle Abbildungen erhalten: werden Sie Mitglied über die Crowdfunding-Plattform Steady und unterstützen Sie damit Urologielehrbuch.de.
Urologielehrbuch.de als Hardcover-Buch
 Aktuell, detailliert und übersichtlich: Urologielehrbuch.de wird auch als hochwertiges Hardcover-Buch veröffentlicht. Die 17. Auflage (Ausgabe 2024) ist seit Oktober 2024 verfügbar, siche Abschnitt Neuigkeiten für die Aktualisierungen und Links für den Buchkauf.
Aktuell, detailliert und übersichtlich: Urologielehrbuch.de wird auch als hochwertiges Hardcover-Buch veröffentlicht. Die 17. Auflage (Ausgabe 2024) ist seit Oktober 2024 verfügbar, siche Abschnitt Neuigkeiten für die Aktualisierungen und Links für den Buchkauf.